“L’anestesia è quella
condizione che rende il paziente insensibile allo stimolo chirurgico” (Oliver Wendell Holmes 1846).
I farmaci anestetici sono
quindi farmaci depressivi che causano la parziale o totale perdita della
sensibilità dolorifica accompagnata da perdita di coscienza. Si tratta di
farmaci con ridotto indice terapeutico (rapporto tra dose letale mediana e dose
efficace mediana) che bisogna maneggiare con molta attenzione.
Gli obiettivi dell’anestesia
sono:
·
Depressione
(globale, ma reversibile) del SNC con perdita della percezione e della risposta
agli stimoli esterni
·
Mantenimento
dell’omeostasi degli organi durante le procedure chirurgiche
·
Minimizzazione
degli effetti potenzialmente deleteri degli anestetici stessi
·
Miglioramento
della ripresa post-operatoria
Le caratteristiche
dell’anestesia generale comprendono:
·
Incoscienza,
depressione dei riflessi ed amnesia (assenza della memoria legata alla fase
intraoperatoria)
·
Analgesia
·
Rilasciamento
muscolare
·
Controllo delle
risposte negative allo stress chirurgico
Un anestetico ideale dovrebbe
garantire un’induzione gradevole e rapida dell'anestesia; una ripresa rapida
dagli effetti del farmaco; un ampio margine di sicurezza; un’assenza di effetti
tossici e collaterali alle dosi utilizzate nella pratica clinica. Tuttavia nessun
anestetico possiede tutte queste caratteristiche quando somministrato da solo;
per questo motivo si ricorre all’utilizzo di più
farmaci per sfruttare le proprietà positive dei singoli e minimizzare gli
effetti collaterali.
Fasi dell’anestesia
(l’anestesista controlla continuamente le funzioni vitali):
·
Premedicazione
o
Fase
caratterizzata da effetti sedativi centrali e di inibizione neurovegetativa,
intesi a facilitare l’anestesia (senza abolizione di coscienza). La
preanestesia serve a controllare l’ansia preoperatoria; prevenire gli effetti
collaterali dell’anestesia (emesi, bradicardia, ipersecrezione salivare);
controllare l’attività nervosa riflessa (pazienti ipertesi o cardiopatici); potenziare
l’azione dei farmaci anestetici; controllare il dolore preoperatorio (nei
pazienti con dolore acuto). È costituita da una associazione di
analgesici narcotici (oppioidi), sedativi (benzodiazepine, barbiturici) o
tranquillanti fenotiazinici (clorpromazina) o α2-agonisti (preanestesia
sedativa), anticolinergici, miorilassanti
·
Induzione
o
È caratterizzata
dai seguenti aspetti: perdita di
coscienza; mantenimento della pervietà delle vie aeree; stabilità
cardiocircolatoria; prevenzione del laringospasmo, vomito e rigurgito. In questa fase il paziente deve essere
condotto al piano anestesiologico ottimale (totale
indifferenza allo stimolo chirurgico con minima o nulla depressione
circolatoria). I farmaci utilizzati comprendono ipnotici, curari, gas freschi (O2, N2O,
aria), oppioidi, antiemetici, cortisonici, gastroprotettori
·
Mantenimento
o
Assicura un
adeguato piano di anestesia al variare dello stimolo chirurgico. È in questa
fase che si realizzano i quattro aspetti fondamentali della anestesia: abolizione dello stato di coscienza (ipnosi);
analgesia; rilasciamento muscolare; controllo della risposta allo stress
chirurgico. I farmaci utilizzati comprendono ipnotici, curari, gas freschi (O2,
N2O, aria), oppioidi, anestetici
inalatori
·
Risveglio
o
È caratterizzato
dal progressivo recupero
neurocomportamentale del paziente, dalla ripresa di un’attività respiratoria spontanea ed
efficace, dalla stabilizzazione
cardiocircolatoria, dal controllo di rigurgito, nausea, vomito, dolore
post-operatorio, brivido. I farmaci utilizzati comprendono gas freschi, antagonisti degli oppiacei e del
curaro, anestetici inalatori (scalati gradualmente), ipnotici (scalati
gradualmente)
·
Dimissione dalla
sala operatoria
I farmaci anestetici si
dividono in due grosse categorie
·
Anestetici per
via parenterale (barbiturici, propofol e chetamina adiuvati dalle
benzodiazepine e/o oppioidi)
·
Anestetici per inalazione
(volatili o gassosi)
L’anestesia generale può
essere definita come una depressione globale ma reversibile della funzione del
SNC con conseguente perdita della risposta a tutti gli stimoli esterni e
annullamento della loro percezione.
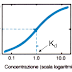
La potenza di un anestetico
generale è di solito misurata determinando la concentrazione che impedisce il
movimento in risposta alla stimolazione chirurgica. Per gli anestetici per via
inalatoria, la potenza anestetica viene misurata in unità MAC (minimum alveolar
concentration), con 1 MAC definito come la minima concentrazione alveolare che
impedisce il movimento in risposta alla stimolazione chirurgica nel 50% dei
soggetti. Si produce anestesia quando la pressione parziale di anestetico
nell’encefalo è uguale o superiore alla MAC. Poiché l’encefalo è molto perfuso
l’induzione dell’anestesia si avrà nel corso di minuti.
I punti di forza della MAC:
·
La concentrazione
alveolare può essere monitorata in modo continuo misurando la concentrazione di
anestetico al termine di un atto respiratorio tramite spettroscopia a raggi
infrarossi o spettrometria di massa
·
Fornisce un
parametro direttamente correlato alla concentrazione libera dell’anestetico a
livello del suo sito di azione nel SNC
·
È un parametro
semplice da misurare che riflette un importante obiettivo clinico
La potenza degli anestetici
per via endovenosa è molto più difficile da misurare. In generale, la potenza
dei composti per via endovenosa è definita come la concentrazione plasmatica
libera (all’equilibrio) che produce perdita della risposta all’incisione
chirurgica nel 50% dei soggetti.
Gli anestetici generali
producono due importanti effetti fisiologici a livello cellulare. In primo
luogo, gli anestetici per inalazione possono iperpolarizzare i neuroni. Secondariamente,
a concentrazioni anestetizzanti gli anestetici per via sia inalatoria sia
endovenosa hanno effetti rilevanti sulla trasmissione sinaptica ed effetti di
entità molto più ridotta sulla generazione o propagazione del potenziale
d’azione.
Diversi canali ionici
ligando-dipendenti, recettori e proteine di trasduzione del segnale sono
modulati dagli anestetici generali. Tra questi, la prova più indicativa di un
effetto diretto degli anestetici si ha per i recettori GABAA e NMDA
e per i canali del K+.
Effetti collaterali dell’anestesia
generale
·
Effetti
emodinamici: l’effetto fisiologico più rilevante dell’induzione dell’anestesia
è una diminuzione della pressione arteriosa sistemica. Tra le cause figurano
una vasodilatazione diretta, una depressione del miocardio, un’attenuazione del
controllo barorecettoriale e una riduzione del tono simpatico centrale. I
diversi composti variano per l’entità dei loro specifici effetti, ma in tutti i
casi la risposta ipotensiva è potenziata a causa della sottostante deplezione
di volume o dalla preesistente disfunzione miocardica. Anche gli anestetici che
mostrano una minima tendenza a causare ipotensione in condizioni normali (es
ketamina) devono essere usati con cautela nelle vittime di traumi, in cui la
deplezione del volume intravascolare è compensata da un’intensa attivazione del
sistema simpatico
·
Effetti
respiratori: quasi tutti gli anestetici generali riducono sia lo stimolo
centrale della ventilazione sia i riflessi che garantiscono la pervietà delle
vie aeree. Pertanto, la ventilazione deve essere generalmente assistita o
controllata almeno per un certo periodo durante la chirurgia. Il riflesso della
stimolazione della faringe è perso e lo stimolo della tosse è attenuato. Anche
il tono dello sfintere esofageo inferiore è ridotto, con possibile rigurgito.
L’intubazione endotracheale è stata una delle principali ragioni della
diminuzione delle morti per aspirazione durante l’anestesia generale. Il
rilassamento muscolare è di enorme importanza durante l’induzione dell’anestesia
generale perché facilita le manovre atte a garantire la ventilazione, tra cui
l’intubazione endotrcheale. Per ottenere tale rilassamento vengono comunemente
utilizzati bloccanti neuromuscolari
·
Ipotermia: i
pazienti sviluppano comunemente ipotermia (<36 °C) a causa della ridotta
temperatura ambientale, della somministrazione endovenosa di fluidi freddi e
dell’alterazione del controllo termoregolatorio
·
Nausea e vomito:
sono causati dall’attività degli anestetici sulla zona chemorecettoriale e sul
centro del vomito del tronco cerebrale. L’uso di propofol come composto di
induzione e del ketorolac (FANS) come sostituto degli oppioidi può ridurre
l’incidenza e la gravità della nausea e del vomito nel periodo post operatorio
Anestetici per via parenterale
Gli anestetici per via
parenterale sono composti idrofobici di ridotte dimensioni. L’idrofobicità è il
fattore chiave che governa la loro farmacocinetica. Dopo un singolo bolo
endovenoso, questi farmaci si ripartiscono preferenzialmente nei tessuti altamente
perfusi e lipofilici dell’encefalo e del midollo spinale, dove producono
anestesia nel tempo di un singolo ricircolo. Successivamente, i livelli ematici
decadono rapidamente, con la conseguente ridistribuzione del farmaco dal SNC al
sangue. L’anestetico si diffonde poi nei tessuti meno perfusi come il muscolo e
gli organi viscerali, e a una velocità minore nel tessuto adiposo poco perfuso
ma molto idrofobico. La cessazione dell’anestesia dopo singoli boli di
anestetico per via parenterale riflette principalmente la ridistribuzione al di
fuori del SNC piuttosto che il metabolismo. La maggior parte della variabilità
individuale nella sensibilità agli anestetici per via parenterale può essere
spiegata dai fattori di tipo farmacocinetico. Per esempio, nei pazienti con
ridotta gittata cardiaca la relativa perfusione cerebrale e la frazione di una
dose di anestetico che raggiunge il cervello sono più elevate; per questo
motivo i pazienti in shock settico e quelli con cardiomiopatia di solito
richiedono dosi ridotte di anestetico. I pazienti anziani tipicamente
richiedono una dose più ridotta di anestetico, soprattutto a causa di un minor
volume iniziale di distribuzione.
L’anestesia per via
parenterale può essere indotta potenziando il sistema gabaergico (benzodiazepine
e barbiturici possono causare anestesia potenziando allostericamente l’azione
del GABA a livello delle sinapsi inibitorie sulle cellule piramidali) o inibendo quello glutammatergico (la ketamina è un
antagonista dei recettori NMDA del glutammato).
Gli anestetici per via
parenterale sono i farmaci più comunemente utilizzati per l’induzione
dell’anestesia del soggetto adulto. La loro lipofilicità, associata alla
relativamente elevata perfusione dell’encefalo e del midollo spinale, comporta
una rapida comparsa dell’effetto e una breve durata d’azione dopo una singola
dose. Tuttavia questi farmaci si accumulano nel tessuto adiposo prolungando la
fase del risveglio se vengono somministrate dosi multiple, in particolare per i
farmaci con ridotta velocità di clearance. Tiopental e propofol sono i due
composti più utilizzati. Il tiopental ha un profilo di sicurezza stabilito da
lungo tempo. Il propofol è vantaggioso per le procedure in cui è desiderabile
un ritorno rapido allo stato mentale normale. L’etomidato di solito viene
riservato ai pazienti a rischio di ipotensione e/o ischemia miocardica, mentre
la ketamina è più indicata per i pazienti con asma e/o per i bambini sottoposti
a procedure dolorose e di breve durata.
Tiopental sodico (Pentothal):
il tiopental sodico è il barbiturico più frequentemente utilizzato per indurre
anestesia. I barbiturici sono forniti come miscele racemiche malgrado
l’enantiomero-specificità della loro potenza anestetica. La miscelazione dei
barbiturici con farmaci in soluzioni acide durante l’induzione dell’anestesia
può comportare una precipitazione del barbiturico come acido libero; per questo
motivo, è pratica comune ritardare la somministrazione di altri farmaci finchè
il barbiturico sia passato attraverso la cannula per la somministrazione
endovenosa. La tipica dose di induzione (da 3 a 4 mg/Kg) di tiopental produce
perdita di coscienza in 10-30 secondi con un effetto di picco a 1 minuto e
durata dell’anestesia da 5 a 8 minuti. I neonati e i bambini richiedono solitamente
una dose di induzione più elevata, mentre i pazienti anziani e le donne in
gravidanza richiedono una dose minore. Le dosi possono essere ridotte dopo
premedicazione con benzodiazepine, oppioidi o agonisti α2-adrenergici, a causa
del loro effetto ipnotico additivo. Il metabolismo (lento) avviene a livello
epatico, l’escrezione a livello renale. La profondità dell’anestesia
riflette le variazioni della concentrazione del tiopentale nel suo sito
d’azione (cervello), che è conseguenza della sua distribuzione: prima al cervello (anestesia profonda primi minuti
dalla somministrazione) e poi ridistribuzione agli altri organi (diminuzione
progressiva dell’anestesia e risveglio). La sua breve durata d’azione è
dovuta alla riduzione della concentrazione plasmatica efficace del farmaco
secondaria alla sua distribuzione nei tessuti e non al metabolismo e
all’eliminazione.
·
Vantaggi
o
Rapido inizio di azione (elevata liposolubilità)
o
Breve durata di azione (ridistribuzione in altri
tessuti)
o
Azione antiemetica
o
Non causa ipertermia maligna
·
Svantaggi
o
L’iniezione perivenosa o arteriosa causa gravi
complicanze locali (dolore acuto e necrosi tissutale; endoarterite e trombosi)
o
Provoca depressione respiratoria, tosse e
laringospasmo e broncospasmo (attenzione in pazienti asmatici), ipotensione in
pazienti ipovolemici
o
Tende ad accumularsi dopo infusione endovenosa
continua (prolungamento del periodo di recupero)
o
Ha azione antianalgesica (necessaria co-somministrazione
con analgesico)
Propofol: è l’anestetico per via parenterale più utilizzato
negli USA. Ha caratteristiche farmacocinetiche simili a quelle del tiopental. A
causa della sua emivita di eliminazione ragionevolmente breve (per la clearance
molto elevata e la lenta diffusione del farmaco dal compartimento periferico a
quello centrale), il propofol è spesso utilizzato per il mantenimento
dell’anestesia oltre che per l’induzione (la rapida clearance comporta una
ripresa più rapida dall’anestesia e permette di allontanare il paziente più
rapidamente dalla sala operatoria una volta risvegliato).
·
Vantaggi
o
Inizio e durata d’azione breve (induzione e
risveglio rapidi con possibilità d’infusione continua)
o
Azione antiemetica
o
Non causa ipertermia maligna
o
Non provoca broncospasmo (va meglio per gli
asmatici rispetto al tiopental)
·
Svantaggi
o
Depressione della respirazione
o
Riduzione pressione arteriosa
o
Dolore nella sede di iniezione
Ketamina: la ketamina ha proprietà uniche che la rendono
utile per anestetizzare pazienti a rischio di ipotensione e broncospasmo e per
certe procedure pediatriche. Tuttavia, marcati effetti collaterali ne limitano
l’impiego di routine. La ketamina produce rapidamente uno stato ipnotico
totalmente distinto da quello degli altri anestetici. I pazienti hanno una
profonda analgesia con perdita della risposta ai comandi e amnesia, ma possono
avere gli occhi aperti, muovere gli arti involontariamente e presentare una
respirazione spontanea. Questo stato catalettico è stato denominato anestesia
dissociativa. Il tempo di comparsa dell’azione dopo una dose endovenosa è
simile a quella degli altri anestetici, ma la durata dell’anestesia di una
singola dose è maggiore.
Etomiodato: l’etomiodato è principalmente utilizzato per
l’induzione dell’anestesia nei pazienti a rischio di ipotensione. Ha una rapida
insorgenza e una breve durata d’azione.
Anestetici per inalazione
Caratteristica degli anestetici generali è quella di causare
un aumento della soglia di eccitazione a cui consegue una diminuzione
dell'attività neuronale, sia spontanea che evocata, in molte regioni del SNC.
Per quanto riguarda gli anestetici inalatori è stato ipotizzato il coinvolgimento
di diversi meccanismi ionici, tra i quali l’attivazione delle correnti di K+.
Gli anestetici inalatori si legano infatti ai lipidi ed alle proteine della
membrana neuronale modificandone la struttura (fluidità) e quindi la
permeabilità agli ioni. Più alta è la liposolubilità dell’anestetico più questo
è in grado di legarsi alla membrana neuronale e, di conseguenza, più è potente
(ma anche tossico).
Un anestetico per svolgere la sua attività deve raggiungere
nel cervello una concentrazione adeguata valutata ed espressa come pressione
parziale. Controllare l’anestesia inalatoria significa, quindi, agire sulla
pressione parziale del gas anestetico del distretto cerebrale. Raggiunto
l'alveolo polmonare con i gas inspirati (il paziente è intubato), il gas
anestetico eserciterà al suo interno una pressione parziale. L'anestetico
tenderà a spostarsi dall’alveolo verso il sangue (ove la sua pressione parziale
è minore o nulla), fino al raggiungimento di un equilibrio fra il distretto
alveolare e quello ematico; in seguito lo spostamento del gas anestetico,
avverrà dal sangue ai tessuti (compreso l’encefalo). Tutte queste pressioni parziali tendono ad equilibrarsi
con quella di volta in volta presente nell’alveolo. La velocità di
trasferimento dell’anestetico dai polmoni al sangue ed ai tessuti è influenzata
dalla sua solubilità. La solubilità è la capacità di un soluto (il farmaco) di
sciogliersi in un mezzo. Essa è espressa come coefficiente di partizione che
descrive come un gas anestetico si distribuisce tra due fasi (gas/sangue) o tra
due solventi (sangue/cervello; sangue/tessuto adiposo) una volta raggiunto
l’equilibrio. Dire che la solubilità
(sangue/gas) dell’isoflurano è 1,4 è come dire che all’equilibrio (ugual
pressione tra i compartimenti analizzati) la concentrazione plasmatica
dell’isoflurano sarà 1,4 volte maggiore della sua concentrazione alveolare.
I composti molto solubili nel sangue vengono rapidamente
"portati via" dal torrente ematico ad ogni sistole e l'equilibrio
(pressione parziale) fra due distretti (alveolo/sangue, sangue/tessuti) si
raggiunge molto lentamente, necessitando (per i lunghi tempi che comporta)
l'erogazione di grandi quantità di anestetico.
La solubilità condiziona anche la velocità di eliminazione e
quindi i tempi di risveglio: per i composti con alta solubilità ematica e
tissutale, le quantità di anestetico accumulate nel tessuto adiposo impediranno
che le pressioni parziali nel sangue (e negli alveoli) scendano rapidamente;
pertanto il risveglio sarà ritardato.
Alotano: è il più potente anestetico generale per
inalazione. È un liquido volatile a temperatura ambiente con un coefficiente di
ripartizione sangue/gas relativamente alto e un elevato coefficiente di
ripartizione sangue/tessuto adiposo. L’induzione con alotano è pertanto
relativamente lenta e la concentrazione alveolare dell’alotano rimane
sostanzialmente più bassa della concentrazione dello stesso nell’aria inspirata
per molte ore dalla somministrazione. Poiché è solubile nel grasso e in altri
tessuti corporei, esso si accumula durante somministrazioni prolungate.
Pertanto, la velocità di risveglio è allungata in funzione della durata di
somministrazione. Per le sue caratteristiche di solito viene utilizzato per il
mantenimento dell’anestesia. Non è pungente ed è pertanto ben tollerato per
l’induzione dell’anestesia per inalazione, impiego più comune nel bambino, nel
quale il posizionamento di un catetere endovenoso nella fase preoperatoria può
essere difficile.
·
Effetti collaterali
o
Depressione diretta del miocardio, riduzione
della pressione arteriosa (ipotensione) in funzione della dose
(dose-dipendenza) e riduzione della gittata e della frequenza cardiaca (attenzione
con i pazienti cardiopatici)
o
Riduzione della ventilazione alveolare ed
effetto broncodilatatore
o
Rilassamento della muscolatura scheletrica
attraverso i suoi effetti deprimenti a livello centrale e il potenziamento dei
miorilassanti (tubocuarina)
o
Ipertemia maligna (mutazione del gene per il
recettore della rianodina Ryr1 che in presenza di alcuni anestetici consente
l’ingresso cellulare di Ca2+): reazione ipermetabolica
incontrollabile della muscolatura scheletrica (grave contrazione muscolare) con
aumento della temperatura corporea, del consumo di O2 e della
produzione di CO2 (se non si interrompe la somministrazione di
alotano può avere esito fatale)
o
Riduce il flusso sanguigno renale (e quindi la
GFR), splancnico ed epatico (possibile riduzione della capacità degli enzimi
microsomiali di metabolizzare i farmaci)
Isoflurano: liquido volatile a temperatura ambiente. Ha un
coefficiente di ripartizione sangue/gas significativamente minore rispetto
all’alotano, di conseguenza l’induzione e il risveglio sono più rapidi.
Inoltre, con l’isoflurano si possono ottenere variazioni della profondità
dell’anestesia molto più rapidamente. È caratteristicamente impiegato per il
mantenimento dell’anestesia dopo l’induzione con altri agenti a causa del suo
odore pungente, ma l’induzione dell’anestesia può essere ottenuta in meno di 10
minuti. È un potente vasodilatatore delle arterie coronarie e produce
contemporaneamente un aumento del flusso ematico coronarico e una riduzione del
consumo di ossigeno a livello miocardico. In teoria, ciò lo rende un anestetico
particolarmente sicuro da usare in pazienti con cardiopatia ischemica. L’isofluorano
è molto più potente dell’alotano nel potenziare i bloccanti neuromuscolari. Riduce
il flusso sanguigno renale (e quindi la GFR), splancnico ed epatico (possibile
riduzione della capacità degli enzimi microsomiali di metabolizzare i farmaci)
e può provocare ipertermia maligna.
Desflurano: liquido altamente volatile a temperatura
ambiente con coefficiente di ripartizione sangue/gas particolarmente ridotto
(0,42) e scarsa solubilità nel tessuto adiposo e negli altri tessuti
periferici. Per questi motivi la concentrazione alveolare (ed ematica) aumenta
rapidamente fino a raggiungere i livelli della concentrazione dell’aria inspirata.
Ciò consente di indurre molto rapidamente l’anestesia e di variare rapidamente
il grado di profondità della stessa modificando la concentrazione nell’aria
inspirata. Anche il risveglio dall’anestesia è molto rapido. È molto utilizzato
nella chirurgia di tipo ambulatoriale. Per le sue caratteristiche irritanti
(può indurre tosse, ipersalivazione e broncospasmo) è somministrato per il
mantenimento dell’anestesia e non per l’induzione. Può provocare ipertermia
maligna.
Sevoflurano: liquido volatile a temperatura ambiente che può
subire una reazione esotermica con la calce sodata disseccata utilizzata per
assorbire l’anidride carbonica, producendo ustioni a livello delle vie aeree
(si deve quindi fare attenzione che il sevoflurano non sia utilizzato con apparecchiature
per anestesia in cui il substrato per l’assorbimento dell’anidride carbonica
sia stato essiccato da un prolungato flusso gassoso). La ridotta solubilità nel
sangue e in altri tessuti consente di indurre rapidamente l’anestesia, di
modificare velocemente il grado di profondità variando la concentrazione
somministrata e di ottenere un rapido risveglio dopo interruzione della
somministrazione. Per queste sue caratteristiche, il sevoflurano è ampiamente
utilizzato, in modo particolare per l’anestesia in regime ambulatoriale. È
indicato per l’induzione dell’anestesia per via inalatoria (in particolare nei
bambini) perché non irrita le vie aeree. Nei bambini è associato a delirium al
risveglio (senza conseguenze a lungo termine). Può provocare ipertermia
maligna.
Protossido d’azoto: N2O
è, a temperatura ambiente, un gas incolore e inodore. È dotato di
attività analgesica (per stimolazione dei neuroni del sistema oppioide nella
sostanza grigia periacqueduttale e dei neuroni adrenergici nel locus coeruleus)
e amnesica; è invece un debole anestetico (antagonista dei recettori NMDA).
Viene principalmente usato per indurre analgesia e lieve sedazione in corso di
interventi odontoiatrici. In sala operatoria viene utilizzato (non puro) in
aggiunta ad altri anestetici per inalazione o per via endovenosa (il protossido
d’azoto riduce sostanzialmente la dose di anestetici per inalazione consentendo
la somministrazione di concentrazioni ridotte di anestetici alogenati). Uno dei
principali problemi del protossido d’azoto è che si scambia con l’azoto in
qualsiasi cavità corporea contenente aria. Inoltre, a causa dei loro differenti
coefficienti di partizione sangue/gas, il protossido d’azoto entra nella cavità
più velocemente di quanto faccia l’azoto nell’uscire, aumentando così il volume
e/o la pressione nella cavità (dovrebbe essere evitato in caso di pneumotorace,
ostruzioni a livello dell’orecchio medio, emboli gassosi, ostruzione di anse
intestinali, bolle d’aria a livello endoculare, bolle polmonari e aria a livello
endocranico).
Adiuvanti per l’anestesia
Benzodiazepine: sebbene siano in grado di produrre
un’anestesia simile a quella dei barbiturici, esse non sono utilizzate per
ipnosi in quanto, a dosi anestetizzanti possono causare amnesia e sedazione
prolungate (l’azione delle benzodiazepine può essere antagonizzata in maniera
selettiva dal flumazenil). Sono ampiamente usati per la premedicazione
anestetica, come coadiuvanti degli anestetici per indurre e mantenere il sonno
e perché inducono amnesia anterograda; sono usate da sole in interventi che non
richiedono analgesia come le endoscopie e il cateterismo cardiaco. Vengono
usate per la prevenzione ed il controllo delle convulsioni indotte da
anestetici locali.
·
Diazepam (Valium) à Dopo iniezione endovenosa viene rapidamente distribuito al
cervello, ma occorrono diversi minuti prima che induca sonnolenza
·
Midazolam (Ipnovel) à è un sale idrosolubile non irritante quando somministrato per
iniezione endovenosa. È dotato di un rapido inizio d’azione, potenza maggiore
del diazepam, rapida eliminazione metabolica (ha quindi un effetto più breve
del diazepam ma l’effetto aumenta con infusioni prolungate) e scarsi effetti
cardiorespiratori (tali caratteristiche lo rendono preferibile alle altre
benzodiazepine in corso di anestesia generale)
Agonisti α2-adrenergici: la clonidina è un farmaco
antiipertensivo con proprietà sedative (mima l’effetto degli oppioidi a livello
del locus coeruleus); riduce la dose necessaria di farmaci analgesici e
anestetici. Somministrata 90 minuti prima dell’intervento chirurgico provoca
sedazione e riduzione dell’ansia. Altri agonisti α2-adrenergici altamente
selettivi a livello centrale usati in anestesia sono l’azapexolo e la
dexmedetomidina.
Analgesici
Ad eccezione della ketamina, nessun anestetico ha
un’efficace azione analgesica. A questo scopo sono utilizzati farmaci in
co-somministrazione con gli anestetici. FANS, inibitori selettivi della COX-2 e
paracetamolo forniscono talvolta un’adeguata analgesia per procedure
chirurgiche minori. Tuttavia, in virtù della rapida e profonda analgesia
prodotta, gli oppioidi sono i principali analgesici usati durante il periodo
perioperatorio. Dosi alte e ripetute (di oppioidi) possono dare depressione
respiratoria e sedazione prolungate per cui è necessaria ventilazione meccanica
(è possibile antagonizzare l’effetto con antagonisti oppioidi come il naloxone
o il naltrexone).
Oppioidi e neurolettici (es Leptofen, fentanil+droperidolo)
sono usati nella neuroleptoanestesia, procedura utilizzata in interventi
chirurgici minori ed in procedure diagnostiche come endoscopie, studi radiologici,
medicazioni di ustioni (buona analgesia, ridotta vigilanza, attività motoria e
contatto con l'ambiente circostante (sedazione), non avviene abolizione dello stato
di coscienza).
Bloccanti neuromuscolari
Determinano il blocco della trasmissione colinergica a
livello della placca neuromuscolare (recettori nicotinici) con conseguente
paralisi flaccida. I curari naturali (tubocurarina, pancuronio, vecuronio) sono
antagonisti competitivi dei recettori nicotinici (N2) della placca
neuromuscolare (l'azione paralizzante inizia dai muscoli oculari e delle mani,
gli ultimi sono i muscoli intercostali e diaframmatici). I curari
depolarizzanti (succinilcolina) agiscono da agonisti nicotinici che depolarizzano
persistentemente la fibra muscolare e quindi la desensibilizzano alla
successiva attivazione da parte dell'ACh.