La
farmacocinetica studia l’evoluzione temporale delle concentrazioni di un
farmaco e dei suoi metaboliti nei diversi fluidi e tessuti dell’organismo. È
quasi sempre identificata dall’acronimo ADME che sta ad indicare
·
Assorbimento
·
Distribuzione
·
Metabolismo
·
Eliminazione
Per
dirla più semplicemente, la farmacocinetica studia ciò che l’uomo fa al farmaco
(si occupa quindi di tutto quello che succede prima che si verfichi un effetto
terapeutico o tossico) ed è utile in quanto ci consente di rispondere a due
domande fondamentali:
·
Qual è la dose
giusta di farmaco da somministrare?
·
Ogni quanto va
somministrata la dose?
Assorbimento: descrive la velocità con cui un farmaco lascia il
sito di somministrazione e l’entità con cui si verifica tale processo.
Il
farmaco come prima cosa deve riuscire ad essere assorbito e, per essere
assorbito, deve passare attraverso delle membrane cellulari. Questo passaggio
può avvenire in modi diversi: filtrazione (sono poche le molecole che
utilizzano questo processo, come acqua, urea e alcol; sostanze idrosolubili
molto piccole); diffusione semplice (i farmaci sono acidi o basi deboli la cui
forma indissociata è quella che attraversa meglio la membrana mediante un
processo di diffusione semplice); diffusione facilitata (sfrutta le proteine transmembrana ed avviene senza
dispendio di energia); trasporto attivo (se
il farmaco è presente in forma dissociata, ionizzata, il suo passaggio richiede
dispendio di energia); fagocitosi/pinocitosi (trasporto vescicolare per proteine e molecole molto grandi).
I
fattori che possono influenzare l’assorbimento dei farmaci comprendono
·
Proprietà del farmaco
o
Peso molecolare
o
Forma della molecola
o
Tipo di formulazione farmaceutica
o
Solubilità nel sito di assorbimento
o
Grado di ionizzazione
o
Liposolubilità relativa della forma ionizzata e non
ionizzata
·
Proprietà
dell’organismo
o
Morfologia e
dimensioni della superficie assorbente
o
Perfusione
dell’area assorbente
o
Razza
o
Età
o
Stato
nutrizionale
o
Stato di salute
·
Fattori esogeni
o
Formulazione
o
Interazione con
altre sostanze
Come regola generale è importante ricordare che la
liposolubilità (e quindi la facilità di attraversare le membrane cellulari) è
maggiore quando il farmaco è presente in forma non ionizzata (in dissociata).
Dalla
formula di Henderson-Hasselback sappiamo che il rapporto tra la forma ionizzata
e quella non ionizzata di un farmaco è uguale a 10pH-pKa. Essendo la
pKa dei farmaci già nota (e facilmente reperibile in internet), questa formula
risulta particolarmente utile se si vuole capire (in base al pH) in quale
distretto verrà assorbito il nostro farmaco: supponiamo di voler somministrare
ad un paziente una benzodiazepina con pKa=3,4; applicando la formula precedente
si evince che, in caso di pH 1,4 (quindi a pH gastrico), la forma prevalente
risulta essere quella indissociata. Ad un pH di 6,4 invece (quindi a pH
intestinale), la forma nettamente prevalente risulta essere quella dissociata.
Dal momento che, come già detto, la forma indissociata risulta essere la più
liposolubile, l’assorbimento di questo farmaco avverrà quindi solo a livello
gastrico e non intestinale. Questa informazione può essere utilissima ad esempio
nel caso in cui si voglia somministrare questo farmaco ad un paziente in
terapia cronica con inibitori di pompa protonica (evento abbastanza frequente):
il farmaco infatti, in questo caso non sortirà alcun effetto (perché alzando il
pH non verrà assorbito) e quindi andrà sostituito.
Distribuzione: rappresenta il processo di ripartizione tra plasma,
liquidi interstiziali e cellulari.
Una
volta assorbito, il farmaco entra nel torrente circolatorio e, attraverso
questo, si distribuisce nell’organismo. I fattori che influenzano la
distribuzione comprendono
·
Caratteristiche fisico-chimiche del farmaco
o
Una volta
somministrato, un farmaco più idrosolubile resterà maggiormente nel sangue, un
farmaco più liposolubile tenderà maggiormente a distribuirsi all’interno delle
cellule (perché più in grado di attraversare le membrane)
·
Fissazione proteica della molecola
o
Per fissazione
proteica si intende il legame farmaco-proteine plasmatiche. La percentuale di
farmaco libero (e quindi farmacologicamente attivo) dipende sia dalla
concentrazione del farmaco sia dalla quota di proteine plasmatiche. È
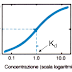
importante notare come, aumentando progressivamente la concentrazione di un
farmaco, l’aumento della quota libera risulti inizialmente lineare per poi
subire una drastica impennata: il drastico aumento della quota libera di
farmaco si verifica nel momento in cui si raggiunge il punto di saturazione di
tutte le proteine plasmatiche. È inoltre importante sottolineare come le variazioni
della quota libera risultino maggiormente evidenti per quei farmaci che
presentano una maggiore fissazione proteica: poniamo ad esempio di essere di
fronte ad un farmaco con un binding proteico del 99%; tutta l’attività
farmacologica, in questo caso, è esercitata dal solo 1% della quantità di
farmaco somministrata, la quota libera. In un caso del genere, piccole
variazioni della quota proteica comportano ben più evidenti variazioni dell’effetto
farmacologico (il passaggio dall’1% al 2% di quota libera ad esempio,
equivarrebbe a raddoppiare la dose somministrata e quindi, potenzialmente, a
trasformare un farmaco efficace in un farmaco tossico). Da queste
considerazioni si può facilmente dedurre che, in caso di pazienti con
ipoproteinemia, è consigliabile utilizzare farmaci con bassa fissazione
proteica
·
Irrorazione degli organi
o
Un farmaco che viaggia nel torrente circolatorio raggiunge prima un
organo ben irrorato rispetto ad uno meno irrorato
·
Affinità specifica dei tessuti
Metabolismo: con questo termine si intende l’insieme delle
reazioni di biotrasformazione di xenobiotici (gli xenobiotici rappresentano
tutte quelle sostanze assunte dall’esterno che non hanno una finalità
nutritiva) per favorirne l’eliminazione dall’organismo.
Il
processo di metabolismo determina quasi sempre la disattivazione del farmaco (cioè
crea effettivamente un prodotto di trasformazione più facile da eliminare) ma
talvolta può provocarne anche l’attivazione (con il termine profarmaco ci si
riferisce ad un farmaco inattivo che, metabolizzato dall’organismo, viene
attivato). Oltre all’inattivazione ed all’attivazione di un farmaco, la fase di
metabolismo può determinarne anche un cambio di attività farmacologica (creare
quindi un metabolita con attività diversa dal farmaco di partenza) o
addirittura, anche se raramente, un aumento della sua tossicità (un esempio di
quest’ultimo caso è rappresentato dal metabolismo epatico del paracetamolo in
alcuni anglosassoni portatori di una particolare mutazione).
L’organo
principalmente coinvolto nel metabolismo dei farmaci risulta essere il fegato,
anche se già a livello del tratto gastrointestinale ci possono essere degli
enzimi che danno il via al processo metabolico. Microscopicamente, le strutture
deputate al metabolismo dei farmaci sono il reticolo endoplasmatico, i
mitocondri ed in parte, i ribosomi. Il complesso enzimatico coinvolto
maggiormente risulta essere quello del citocromo P450, in particolare CYP3A4 (metabolizza
circa il 30-50% dei farmaci). Le reazioni metaboliche vengono tradizionalmente
classificate in reazioni di fase 1 (o reazioni di introduzione di un gruppo
funzionale o reazioni di funzionalizzazione) e reazioni di fase 2 (o reazioni
di coniugazione). Non tutti i farmaci passano prima attraverso una reazione di
fase 1 e poi una di fase 2, per questo risulta meglio parlare di reazioni di
funzionalizzazione e di coniugazione (i termini fase 1 e fase 2 danno il senso
di un ordine temporale che in realtà non esiste). Alcuni farmaci possono
inoltre subire un (ri)circolo entero-epatico e quindi essere riconvertiti,
successivamente alla coniugazione epatica, in farmaco attivo. Questa eventualità
determina un allungamento temporale dell’effetto farmacologica, fattore che
influenza molto la posologia.
I
fattori che influenzano la biotrasformazione dei farmaci sono:
·
Inibizione degli enzimi metabolizzanti
·
Induzione degli enzimi metabolizzanti
·
Polimorfismi genetici
·
Patologie (ad esempio in caso di epatopatia il metabolism
epatico può risultare diminuito e la tossicità, di conseguenza, aumentata)
·
Età (ad esempio molti neonati nascono con deficit di
citocromi)
·
Sesso (alcuni citocromi risentono della produzione
ormonale)
·
Interazioni farmaco-metaboliche
Eliminazione: l’eliminazione è l’ultima fase della farmacocinetica
e descrive tutti quei processi mirati all’escrezione del farmaco, o del suo
metabolita, dall’organismo.
Le vie
di eliminazione di un farmaco comprendono
·
Vie principali
o
Renale
§ È la via di eliminazione preferita. Essendo l’urina, come il sangue, composta
principalmente da acqua, i composti che più facilmente vengono eliminati per
via renale sono quelli idrosolubili. Se per l’assorbimento di un farmaco,
quindi, doveva prevalere la forma indissociata, per la sua eliminazione deve
prevalere invece la forma dissociata
o
Epatica
§ Quando il PM supera i 500 KDa e/o il metabolita non è
idrosolubile, l’eliminazione avviene attraverso la bile e, quindi, le feci
·
Vie secondarie
o
Polmonare
o
Intestinale
o
Cutanea (col
sudore)
o
Salivare
o
Lacrimale
o
Mammaria (diventa
critica durante l’allattamento)