Per farmaco si
intende ogni sostanza capace di provocare in un organismo modificazioni
funzionali mediante un’azione chimica o fisica. I farmaci, nella gran parte dei
casi, agiscono legandosi a recettori (anche se non sempre, ad esempio l’acqua
ossigenata ha proprietà antibatteriche in quanto promuove la formazione di
ROS). Un farmaco quindi non crea un effetto, ma modula una funzione
preesistente alterando lo stato funzionale del suo recettore. Per recettore si intende qualsiasi molecola
la cui funzionalità è modificata dall’interazione con un farmaco.
Per molti farmaci, quindi, l’attività biologica è dovuta
alla formazione di un complesso con i loro recettori:
R+X=RX
(dove R sta per recettore ed X per farmaco)
L’interazione tra farmaco e recettore è generalmente mediata
da legami chimici deboli (legami ionici, ponti idrogeno, attrazioni di van der
Waals, interazioni idrofobiche). Perché il legame consenta di generare un
effetto biologico, è necessario che
·
Il numero di legami a bassa energia sia relativamente
elevato (un numero estremamente elevato di legami determina la formazione di
legami irreversibili, come ad esempio quello tra l’alfa-bungarotossina di cobra
ed il recettore nicotinico per l’acetilcolina)
·
Le superfici di farmaco e molecola siano
complementari (ed il sito di legame recettoriale sia esposto)
o
Bisogna ricordare però che il farmaco può
riconoscere superfici molecolari complementari presenti su macromolecole
diverse dal suo “recettore” principale. È questo il caso ad esempio dei
broncodilatatori β2 agonisti, i quali sono in grado di legarsi ai recettori β2
presenti a livello della muscolatura bronchiale ma anche, a dosi più elevate,
ai recettori β1 adrenergici presenti nel tessuto cardiaco. Questo fenomeno è
spiegato dal fatto che la complementarietà del farmaco è massima per i
recettori β2 e solo parziale per i β1 (per aumentare la probabilità di
interazione farmaco-recettore, in questo secondo caso, sarà necessario quindi
aumentare la concentrazione del farmaco). La selettività di un farmaco quindi,
è funzione non solo della complementarietà farmaco-recettore, ma anche dalla
concentrazione del farmaco stesso
I principali parametri che caratterizzano l’interazione di
un farmaco con i suoi siti di legame sono
·
Costante di
dissociazione à
risulta dal rapporto tra le concentrazioni di recettore e farmaco liberi all’equilibrio
e la concentrazione del complesso farmaco-recettore all’equilibrio; è quindi indice
dell’affinità di un farmaco per il suo recettore (è inversamente correlata all’affinità
del ligando per il recettore: tanto più è alta Kd tanto minore sarà la probabilità che il farmaco risulti
legato al proprio sito recettoriale)
o
Kd= [R] [X]/[RX] = 1/Ka
·
Densità dei
siti di legame à
generalmente indicata con Bmax
o RT, per densità dei siti si intende il numero massimo di siti di
legame presenti per cellula
·
Costanti cinetiche à sono le costanti di
velocità della reazione diretta (formazione del complesso), Kon, e
inversa (scissione del complesso), Koff, per cui vale la relazione
o
Koff/Kon=Kd
La relazione tra concentrazione di farmaco e complesso
farmaco-recettore è simile all’equazione di Michaelis-Menten (che descrive l’interazione
enzima-substrato):
B= [X] Bmax/Kd+Z
La concentrazione di farmaco legato al recettore (B=[RX])
dipende direttamente dal numero massimo di recettori (Bmax) e dalla
concentrazione di farmaco ([X]) ed inversamente dalla costante di dissociazione
(quanto il farmaco è in grado di legare il recettore) corretta per un
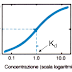
coefficiente (Z). Graficamente, l’isoterma
di legame (o isoterma di Langmuir), è esprimibile in scala semilogaritmica come
segue:

L’andamento sigmoidale è indicativo del fatto che, oltre una
certa concentrazione di farmaco, ad un aumento di concentrazione non
corrisponde un aumento dell’effetto del farmaco stesso, in quanto si è raggiunto
il livello di saturazione di tutti i recettori. L’isoterma di legame e le sue trasformazioni
lineari permettono di ricavare i parametri dell’interazione farmaco-recettore.
Quanto detto finora è valido nel caso in cui il ligando in
esame interagisca con un solo recettore. In presenza di sottotipi recettoriali
multipli che differiscano tra loro per affinità e/o per numero, le curve di
interazione farmaco-recettore risultano modificate. Inoltre farmaci diversi
possono competere per il legame a uno stesso recettore: la competizione determina
uno spostamento delle curve di legame e quindi una riduzione apparente dell’affinità.
La relazione fra concentrazione di un farmaco e il grado di
risposta ottenuto prende il nome di curva concentrazione-risposta. Quando la
sperimentazione è condotta in vivo, l’effetto è messo in relazione con la dose
di farmaco somministrata e la relativa curva è chiamata curva dose-risposta. Le risposte farmacologiche possono essere
graduali e misurabili in continuo (ad esempio l’aumento di pressione
sanguigna); non classificabili in continuo ma ordinabili con uno score o uno
stage (ad esempio il grado di dolore) oppure di tipo tutto o nulla (ad esempio
morte o remissione completa di malattia).
Sulla base della curva dose-risposta si possono definire
alcuni parametri fondamentali
·
Potenza
à
la potenza di un farmaco è definita sulla base della dose necessaria per
ottenere un determinato effetto: più una curva è situata a sinistra sull’asse
delle ascisse, più potente è il farmaco a cui la curva si riferisce. Questo parametro
influenza quindi la dose necessaria per ottenere un determinato effetto,
fattore da tenere in considerazione ad esempio in pazienti sottoposti a
politerapia (in questi casi può essere consigliato preferire un farmaco potente
in modo da minimizzare la dose somministrata). Per convenzione si assume come
valore di paragone della potenza quella concentrazione di farmaco che genera un
effetto pari al 50% dell’effetto massimo (effective concentration 50, EC50)
o, in caso di somministrazione in vivo, quella dose che produce il 50% dell’effetto
massimo (effective dose 50, ED50)
·
Efficacia
à l’efficacia
di un farmaco è definita sulla base dell’entità massima dell’effetto che il
farmaco può indurre

Potenza ed efficacia non sono quindi termini
intercambiabili, al contrario è molto importante ricordarne le differenze. Un
esempio utile è rappresentato dai diuretici: i diuretici tiazidici sono i più
potenti (attivi a basse concentrazioni); i diuretici dell’ansa (furosemide)
sono invece i più efficaci (dotati dell’effetto diuretico massimo).
Agonisti, antagonisti e modulatori
È definito agonista
un farmaco che si lega ad un recettore in modo tale da generare una risposta
biologica.
Un antagonista
recettoriale è definito come un farmaco che legandosi ad un recettore
inibisce (parzialmente o completamente a seconda della concentrazione) l’effetto
di un agonista che agisca attraverso lo stesso recettore.
Altri tipi di antagonisti (che tuttavia non sono definibili
come antagonisti in senso stretto) sono gli antagonisti funzionali, a loro volta
divisi in fisiologici (un antagonista funzionale fisiologico consiste in un
agonista che produce un effetto contrario a quello di un altro farmaco e quindi
ne inibisce l’azione pur non interagendo con lo stesso recettore) ed indiretti
(un antagonista funzionale indiretto consiste in un inibitore di una molecola
intermedia tra il recettore e il suo effetto finale).
Sullo stesso recettore possono coesistere più siti di
legame: quello ortosterico è il sito occupato dall’agonista endogeno, qualsiasi
altro sito è invece definito allosterico. Se il legame tra agonista e
antagonista è mutualmente esclusivo, l’antagonismo è competitivo. Gli antagonisti
competitivi possono produrre un effetto reversibile o irreversibile; nel primo
caso sarà modificata la potenza del farmaco agonista ma non la sua efficacia
(la curva dose-risposta viene spostata parallelamente verso destra), nel
secondo caso invece sarà modificata l’efficacia del farmaco agonista ma non la
sua potenza (la curva dose-risposta viene depressa ma non traslata).
I farmaci possono anche legarsi a siti allosterici ed essere
quindi definiti modulatori allosterici.
I modulatori allosterici vengono a loro volta divisi in antagonisti allosterici
(possono agire come gli antagonisti competitivi irreversibili, deprimendo la
curva dose-risposta, oppure possono determinare una traslazione e contemporanea
depressione della curva, riducendo quindi sia la potenza che l’efficacia del
farmaco), potenziatori allosterici (aumentano l’affinità e/o l’efficacia di un
farmaco ortosterico) ed agonisti o attivatori allosterici (attivano il
recettore da soli, senza la necessità di un legame agonista-recettore ortosterico,
legandosi a un sito allosterico).
Nella maggioranza dei casi tuttavia, i farmaci sono agonisti parziali, ossia possiedono
caratteristiche intermedie tra quelle di un agonista e quelle di un antagonista.
Agonisti, antagonisti ed agonisti parziali sono definibili
anche sulla base della cosiddetta attività intrinseca: si definisce attività
intrinseca (α) la capacità del farmaco di dare inizio alla risposta biologica,
una volta che esso si sia legato al recettore. L’attività intrinseca può
assumere valori compresi fra 0 e 1; più precisamente, per un agonista α=1; per
un antagonista α=0 e per un agonista parziale 0<α<1. Quando tutti i
recettori presenti sono occupati da un agonista parziale si ottiene un effetto Δ=αΔmax.
In realtà si verifica spesso il caso in cui la curva
dose-risposta si trova a sinistra della curva di interazione farmaco-recettore
(ovvero la concentrazione alla quale si raggiunge la metà dell’effetto è minore
della concentrazione alla quale risulta legata metà della dose di farmaco
somministrato; EC50<Kd). Esistono diverse possibilità
per spiegare il fatto che la curva dose-risposta sia a sinistra della curva di
binding, una di queste è la presenza di un sistema di amplificazione a cascata.
Secondo la teoria classica, il complesso farmaco-recettore
(RX) è l’unica entità in grado di generare una risposta biologica. Agli inizi
degli anni ’90 tuttavia, sono stati scoperti recettori (soprattutto accoppiati
a proteine G) in grado di generare la risposta anche in assenza di ligando; i
recettori costitutivamente attivi. Secondo la teoria dell’attivazione
costitutiva, i recettori esistono naturalmente in almeno due stati possibili, R
(inattivo) ed R* (attivo), in equilibrio tra loro (in condizioni
basali l’equilibrio risulta spostato nettamente verso R). Nell’ambito di questa
teoria, si definisce agonista il farmaco che mostra un’affinità per R* maggiore
che per R: la formazione del complesso R*X sarebbe quindi favorita; R* verrebbe
sottratto dall’equilibrio e ciò indurrebbe una trasformazione di R in R*. Viceversa, si definisce agonista inverso
il farmaco che presenta maggior affinità per R che per R*. Gli agonisti inversi
sono gli unici farmaci in grado di diminuire l’attività basale di un sistema:
la loro attività intrinseca α sarà quindi compresa tra 0 e -1.